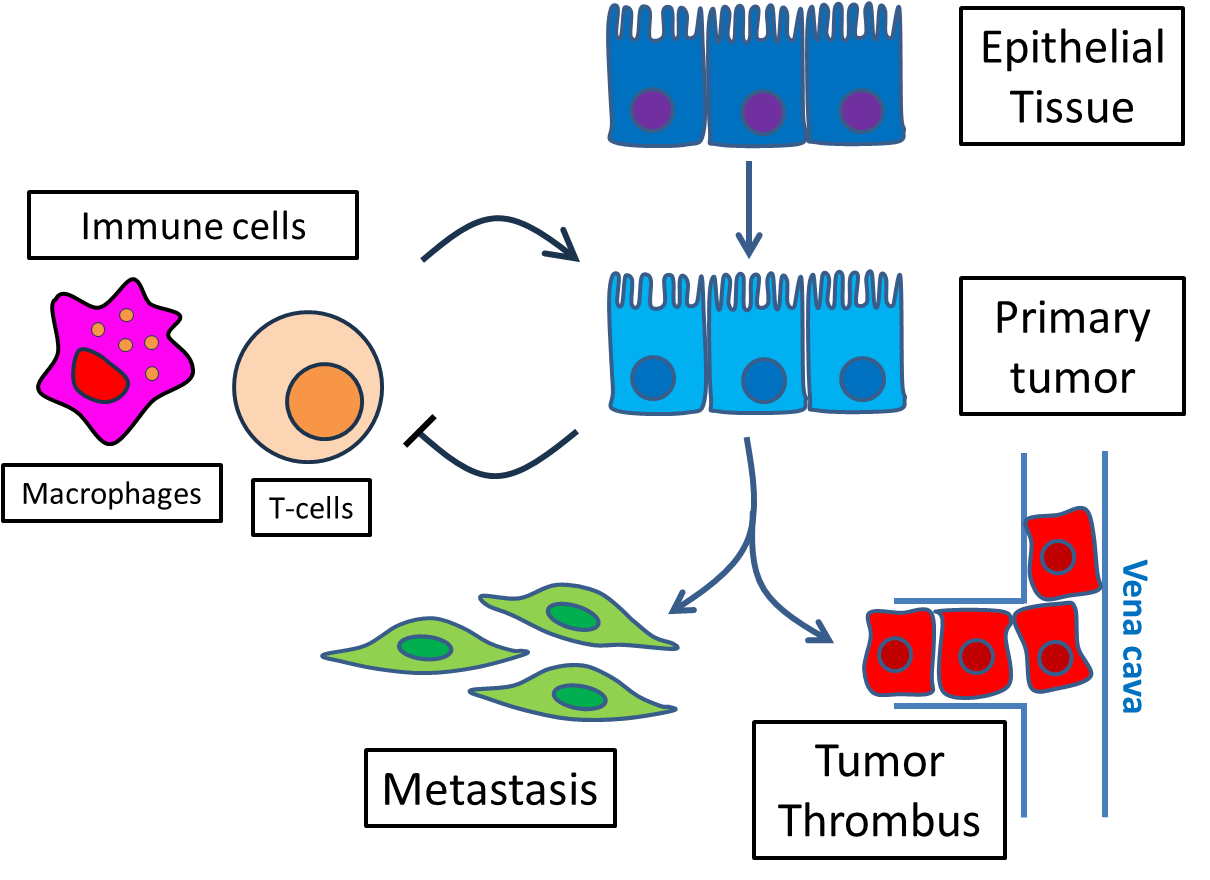
A tissue or tumor is a complex system composed of thousands of interacting cells. Within each cell there is a regulatory network of genes in which transcription factors bind to enhancer regions in the DNA and activate or inhibit target genes.
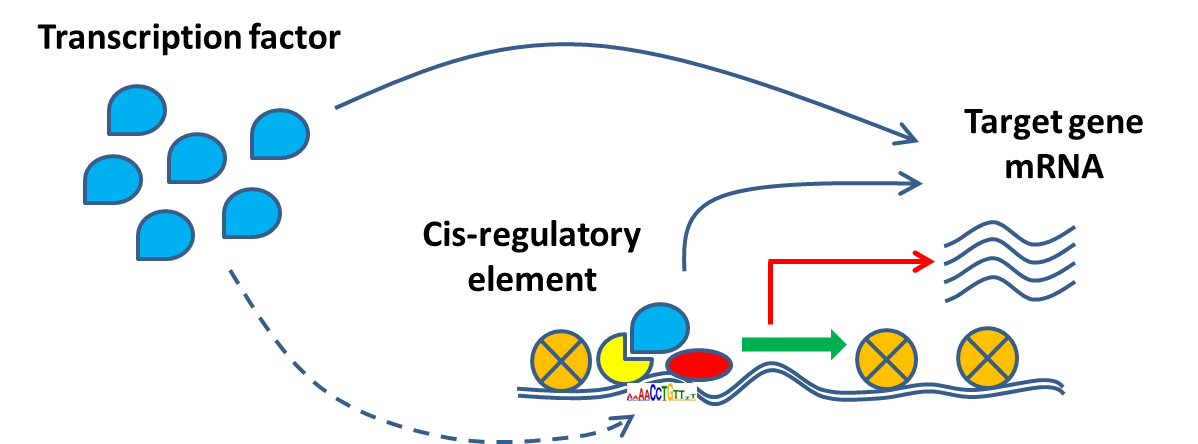
In our lab we use statistical inference algorithms to reconstruct gene regulatory networks from single-cell multiomics data, that is, simultaneous profiling of RNA expression and chromatin accessibility in thousands of single cells or nuclei. The network model assumes that a transcription factor regulates a target gene by binding to cis-regulatory elements in its vicinity if the following criteria are met:
(1) The expression levels of the transcription factor and its target gene are correlated or anti-correlated.
(2) There are putative enhancer regions located near the target gene, that is, accessible chromatin domains that are enriched for DNA binding motifs of the transcription factor.
(3) There is a significant correlation or anti-correlation between the accessibility counts of the putative enhancer regions and the RNA expression counts of the target gene.
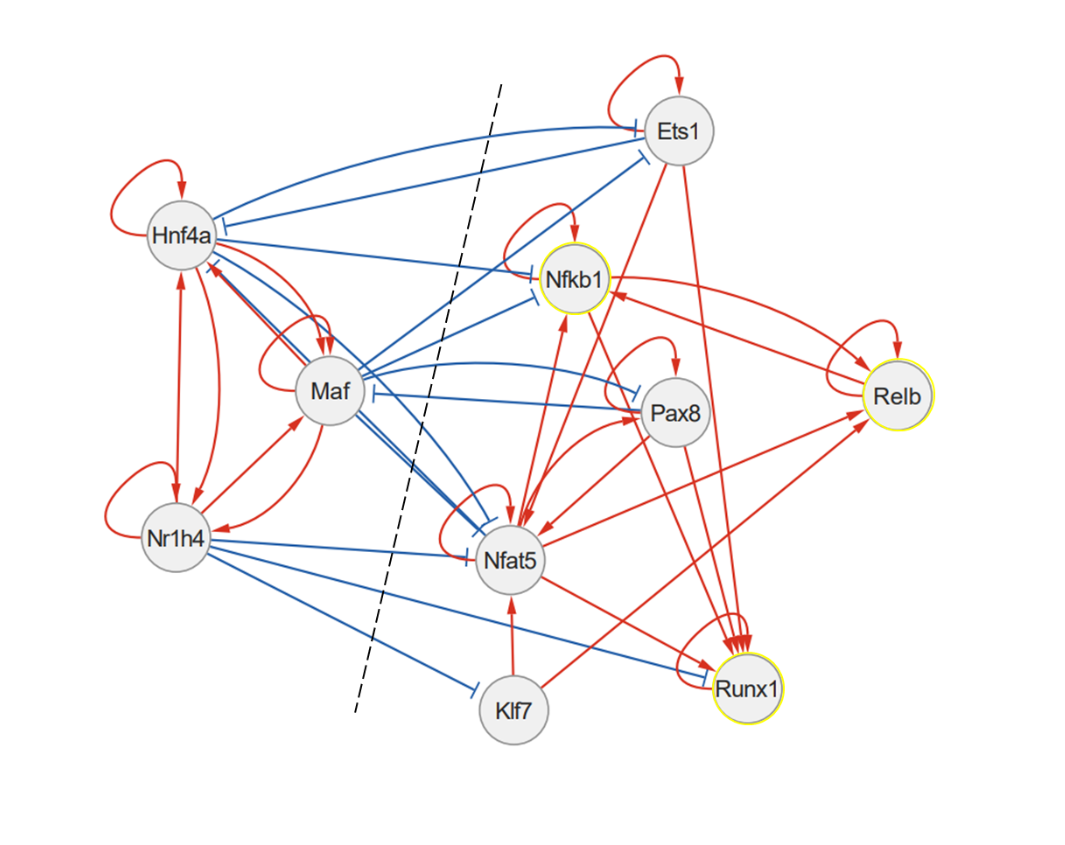
A “regulon” consists of a transcription factor along with its associated binding sites on the DNA and corresponding target genes. By combining all regulons together, we construct a gene regulatory network (GRN).
Within each network, we aim to find modules of functionally interconnected regulons (“communities”) that control specific biological functions or determine cell phenotype. We then simulate gene expression dynamics for each of these modules to gain insight into how cell states in tissues and tumors change over time and in response to external cues.
Our vision is to use GRNs inferred from patient tumors to simulate anti-cancer therapies and test their efficacy before applying them to patients.