Welcome to the Samson lab!
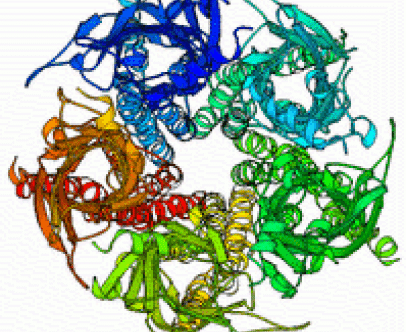
Normal modes dynamics of the acetylcholine receptor and its complex with snake toxins
Calculating protein dynamics is essential for a better understanding of many biological processes. To better understand the motion involved in channel opening of the acetylcholine receptor (AChR), we calculated the normal mode dynamics of the AChR. The normal modes calculations reveal a twist like gating motion responsible for channel opening. This motion is shown in the movie on the right. Throughout this twist motion, the ion channel diameter is shown to increase. Strikingly, the twist motion and the increase in channel diameter are not observed for the acetylcholine receptor in complex with two α-bungarotoxin (αBTX) molecules. The toxins seem to lock together neighboring receptor subunits thereby inhibiting channel opening. Interestingly, one αBTX molecule is sufficient to prevent the twist motion. These results shed light on the gating mechanism of the acetylcholine receptor, and present a complementary inhibition mechanism by snake venom derived α-neurotoxins.
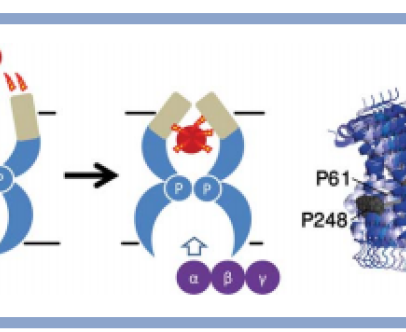
Elastic network normal mode dynamics reveal the GPCR activation mechanism
G-protein-coupled receptors (GPCR) are a family of membrane-embedded metabotropic receptors which translate extracellular ligand binding into an intracellular response. Here, we calculate the motion of several GPCR family members such as the M2 and M3 muscarinic acetylcholine receptors, the A2A adenosine receptor, the β2 -adrenergic receptor, and the CXCR4 chemokine receptor using elastic network normal modes. The normal modes reveal a dilation and a contraction of the GPCR vestibule associated with ligand passage, and activation, respectively. Contraction of the vestibule on the extracellular side is correlated with cavity formation of the G-protein binding pocket on the intracellular side, which initiates intracellular signaling. Interestingly, the normal modes of rhodopsin do not correlate well with the motion of other GPCR family members. Electrostatic potential calculation of the GPCRs reveal a negatively charged field around the ligand binding site acting as a siphon to draw-in positively charged ligands on the membrane surface. Altogether, these results expose the GPCR activation mechanism and show how conformational changes on the cell surface side of the receptor are allosterically translated into structural changes on the inside
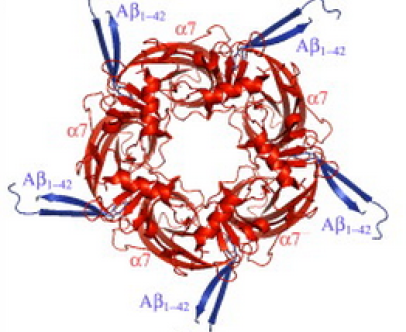
Modeling the binding mechanism of Alzheimer's Aβ1-42 to nicotinic acetylcholine receptors based on similarity with snake α-neurotoxins
For over a decade, it has been known that amyloid β (Aβ) peptides of Alzheimer's disease bind to the nicotinic α7 acetylcholine receptor (AChR) with picomolar affinity, and that snake α-neurotoxins competitively inhibit this binding. Here we propose a model of the binding mechanism of Aβ peptides to α7-AChR at atomic level. The binding mechanism is based on sequence and structure similarities of Aβ residues with functional residues of snake α-neurotoxins (ATX) in complex with AChR. The binding mechanism involves residue (Aβ)K28 (similar to (ATX)R32) which forms cation/π interactions in the acetylcholine binding site, and residues (Aβ)G29-(Aβ)I32 [GAII] (similar to (ATX)G33-(ATX)I36 [GTII]) which form an intermolecular β-sheet with residues (α7)F189-(α7)E191 of AChR. Through these interactions, we propose that the AChR serves as a chaperone for Aβ conformational changes from α- to β-hairpin. The interactions which block channel opening provide fundamental insight into Aβ neurotoxicity and cognition impairment, that could contribute to pathogenic processes in Alzheimer's disease, thus paving the way for structure based therapies
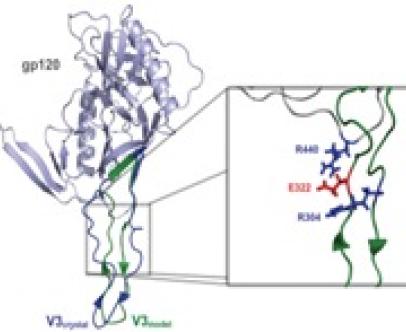
Modeling of Protein Structure: The surface protein gp120 of the HIV virus
Much information and insigth at atomic resolution can be gained from the 3D structure of protein models. In the past, we constructed a model of the gp120 of R5 HIV with the hypervariable V3 loop using homology modeling. The model is based on NMR data for the V3 loop and the crystal structure of the gp120-core. The model is also based on interactions revealed by sequence analysis, i.e. the correlated mutations at positions 322 and 440, suggesting that these two residues interact closely. This model contributes to our understanding of biological phenotype conversion of HIV.
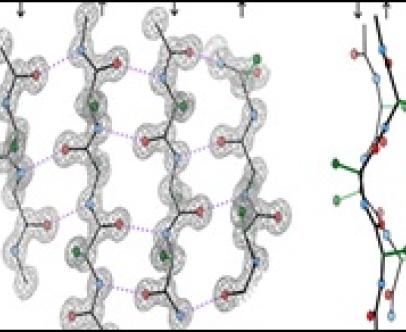
Prediction of residue pairing in beta-sheets
Amino acid propensities for α-helix and β-sheet formation have long been known. Recently, I calculated the propensities of residue pairing in β-structure (unpublished results). This propensity is capable of predicting pairing of residues across &beta-sheets as well as β-bulges.
PSF: an online search engine for protein segment motifs in the PDB
To assist the task of finding related conformations in the Protein Data Bank (PDB), I designed a search engine named protein segment finder (PSF) (1). The search engine uses a compact database to quickly identify protein segments obeying a set of primary, secondary and tertiary structure constraints. The database contains information such as amino acid sequence, secondary structure, disulfide bonds, hydrogen bonds, and atoms in contact as calculated from all protein structures in the PDB. The search engine parses the database and returns hits that match the queried parameters. PSF which is notable for its high speed and interactive feedback, is expected to assist scientists in discovering conformation homologs and predicting protein structure. PSF is publicly available online at http://ari.stanford.edu/psf